
Tablets are solid drug delivery system prepared by compressing a single dose of one or more active drug substance(s) with some additives/ pharmaceutical excipients. They may be circular, oblong, oval, triangular or cylindrical in shape and flat-, round-, concave- or convex-faced with straight or bevelled edges.
In tablet formulation development and during manufacturing of tablet dosage forms, a number of quality control tests are performed to ensure that tablets produced meet the requirements as specified in official compendium and conventional requirements established by the industries over the years. These tests can be grouped into two broad categories namely:
- Pharmacopoeial or Official tests
- Non-pharmacopoeial or Non-official tests
Pharmacopoeial or Official tests
They are called official tests because the test methods are described in official compendia such as the British Pharmacopoeia, American Pharmacopoeias etc. They are standardized test procedures which have clearly stated limits under which compressed tablets could be accepted. These tests include:
- Content of Active Ingredient/ Absolute drug content test
- Uniformity of Weight
- Uniformity of Content
- Disintegration time test
- Dissolution test
These tests are traditionally concerned with the content and the in vitro release of the active ingredient. It must be emphasized that what is presented here should by no means replace what are presented on each of the tests in official compendia.
Content of Active Ingredient
This is determined from a sample of 20 tablets which should be randomly selected from a batch of tablets. The tablets are weighed together and are crushed in a mortar with a pestle.
An amount equivalent to the theoretical content of each tablet or the average of the crushed tablets is weighed out in an analytical balance. The weighed powder is dispersed in a solvent in which the active drug is freely soluble or in a solvent prescribed in the individual drug monograph.
This is filtered and an aliquot of the resultant filtrate is subjected to the stipulated assay procedures. The assay procedures are usually given in the individual drug monograph.
Analysis of the active drug is usually carried out using spectrophotometry or High-Performance Liquid Chromatography (HPLC). The formulation scientist must be familiar with Beer-Lambert’s law. This could be found in relevant analytical textbooks.
It must be emphasized that the results obtained here gives the average content of 20 tablets but does not give indication of the variation of drug content among the individual tablets. The limits of acceptance or rejection of tablets batches are usually presented in the individual drug monograph.
Uniformity of Weight/ Weight variation test
The test for uniformity of weight is performed by weighing individually 20 tablets randomly selected from a tablet batch and determining their individual weights. The individual weights are compared with the average weight.
The sample complies with USP standard if no more than 2 tablets are outside the percentage limit and if no tablet differs by more than 2 times the percentage limit.
Coated tablets are exempted from these requirements but must conform to the test for content uniformity.

Uniformity of weight is a function of granulation quality, flow of granulation and machine performance. However, sometimes these ranges are not sufficiently narrow.
Uniformity of Content
Content uniformity test was developed to ensure content consistency of active drug substances within a narrow range around the label claim in dosage units. This test is crucial for tablets having a drug content of less than 2 mg or when the active ingredient comprises less than 2% of the total tablet weight.
By the USP method, 30 tablets are randomly selected, 10 of these tablets are assayed individually according to the method described in the individual monograph. Unless otherwise stated in the monograph, the requirements for content uniformity are met if the amount of active ingredient in nine (9) of the ten (10) tablets lies within the range of 85% to 115% of the label claim. The tenth tablet may not contain less than 75% or more than 125% of the labelled drug content.
If one or more dosage units do not meet these criteria, the remaining 20 tablets are assayed individually and none may fall outside of the 85% to 115% range for the batch to be accepted.
Various factors are responsible for the variable content uniformity in tablets. This may include:
- Tablet weight variation.
- Uneven distribution of the drug in the powder or granules
- Segregation of the powder mixture or granulation during formulation processes
Disintegration Time Test
For tablets, the first important step towards drug dissolution is breakdown of the tablets into granules or primary powder particles, a process known as disintegration. All USP tablets must pass a test for disintegration, which is conducted in vitro using a disintegration test apparatus.

Disintegration test apparatus
The apparatus consists of a basket-rack assembly containing six open-ended transparent tubes of USP-specified dimensions, held vertically upon a 10-mesh stainless steel wire screen.
During testing, a tablet is placed in each of the six tubes of the basket, and through the use of a mechanical device, the basket is raised and lowered in a bath of fluid (e.g. water, or as prescribed in the individual drug monograph) at 29 to 32 cycles per minute, the wire screen always below the level of the fluid. For most normal release tablets, the time permitted is 15 minutes.
Tablets are said to have disintegrated if no fragments (other than fragments of coating) remains on the screen, or if particles remain, they are soft without an unwetted core. Chewable tablets are not required to comply with the test.
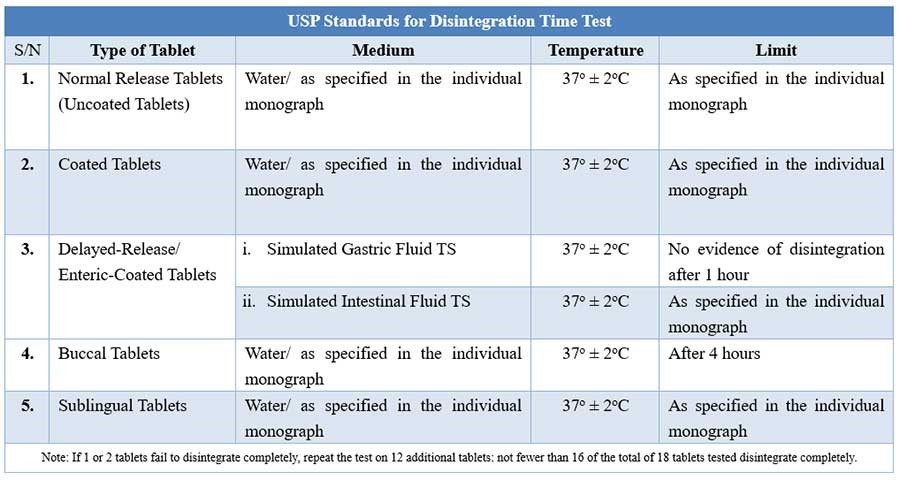
USP Disintegration testing condition and interpretations
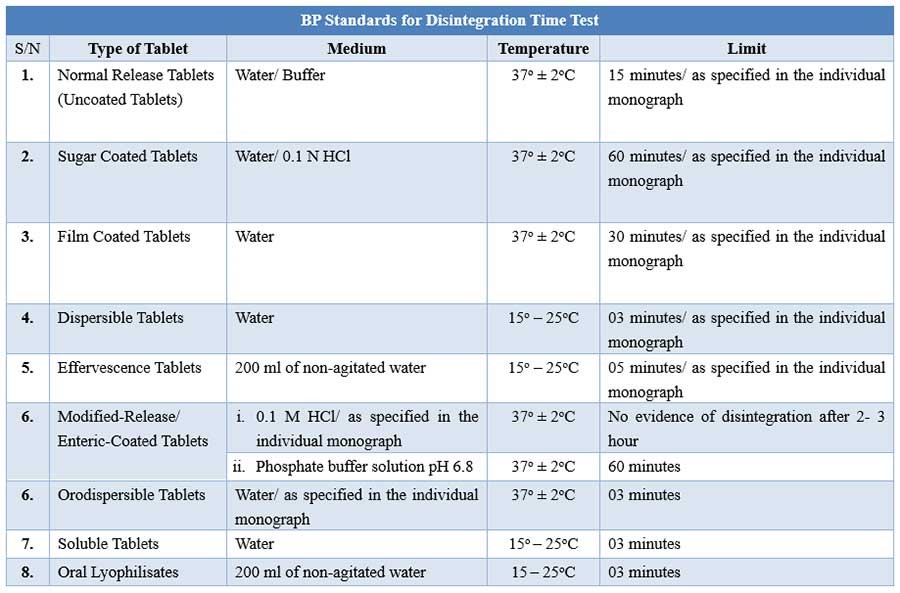
BP Disintegration testing condition and interpretations
Research has established that one should not automatically expect a correlation between disintegration and dissolution. However, since the dissolution of drug from the fragmented tablet appears to control partially or completely the appearance the drug in the systemic circulation, disintegration is still used as a guide by the formulator in the preparation of an optimum tablet formula and as an in-process control test to ensure batch to batch uniformity.
Factors affecting disintegration of tablets include:
- Medium used
- Temperature of the test media
- Operator’s experience
- Nature of the drug
- The diluent used in the formulation
- The type and concentration binder used
- Type and amount of disintegrant used including method of incorporation.
- The presence of excessive lubricants and overly mixed lubricants
- Compressional force used.
Dissolution Test
This test measures the amount of time required for a given percentage of the drug substance in a tablet to go into solution under a specified set of conditions. It is intended to provide a step toward the evaluation of the physiological availability of the drug substances.
In vitro dissolution test is performed using a variety of equipment/apparatus. The British Pharmacopoeia recommends three types of apparatus – the rotating basket, the rotating paddle and the flow-through cell. The static-basket magnetic stirrer assembly can also be used for this test.
The rotating paddle method is generally more discriminatory than the basket method. The flow-through cell method is very useful particularly for
- Poorly soluble active constituents (can use large volume to achieve sink conditions)
- Enteric-coated products (can easily change between different pH fluids)
- Modified release products.

USP Dissolution Apparatus 2
The dissolution medium for each drug is available in the individual drug monograph. For basic drugs, acidic media are used (e.g. 0.1 M hydrochloric acid) while alkaline media are used for acidic drugs (e.g. alkaline buffers). For drugs with non-ionizing molecules, water is recommended.
Dissolution rate test is performed at 37 ± 1 oC. Samples are removed from the dissolution chamber at periodic intervals and analyzed for drug content using a spectrophotometer. Dissolution samples removed for assay should be filtered to remove particles of drugs present, and to exclude tablet excipients that might otherwise interfere with the assay. Non-absorbent filter papers are recommended.
Most commonly, the results of dissolution tests are expressed in terms of the time required to release some percentage of labelled amount of drug from the dosage form. This approach is reported to be particularly useful for quality control purposes once the dissolution characteristics of a drug and dosage form are well understood.
For tablet dosage form design purposes, and for critical product comparison, however, the time required for substantially complete 80 to 90% release or amount released versus time profiles are the most desired approach.
While in vitro dissolution experiment may not correlate perfectly with in vivo bioavailability, the concept of dissolution efficiency proposed by Kahn and Rhodes could be employed to assess the most probable in vivo performance of a tablet formulation.
Dissolution test is not designed to measure the efficacy or safety of the tablet being tested. Both the effectiveness and safety of a specific dosage form must be demonstrated, initially, by means of appropriate in vivo studies and clinical evaluation.
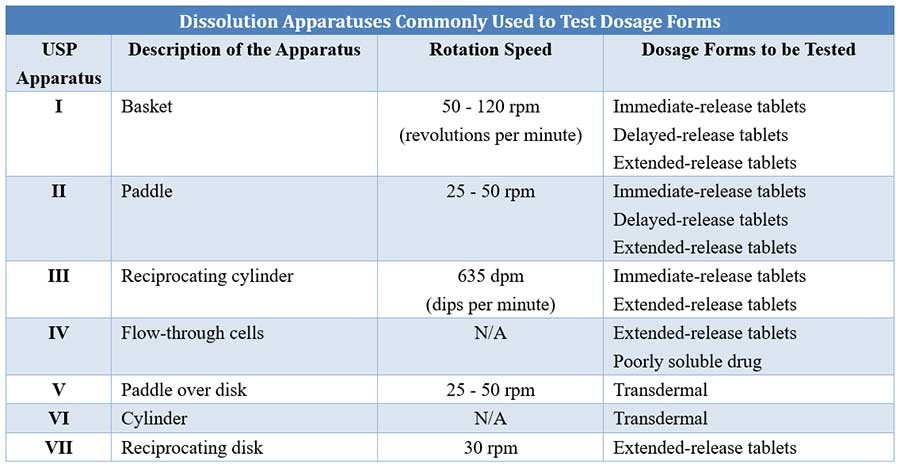
Comparison of different USP dissolution apparatuses
Various factors can affect the dissolution of a drug; they are classified under three categories as follows:
1. Physiochemical properties of the drug
- Polymorphic form: A metastable form of a solid has higher solubility and dissolution compared to its stable counterpart.
- Particle size: The smaller the particle size of a solid, the larger the particle surface area and the higher the dissolution.
- Salt form: A salt form of a drug has a higher aqueous solubility compared to its conjugate acid or base, as well as higher dissolution.
- Hydrates versus anhydrates: The anhydrous form shows higher dissolution than hydrates due to their solubility differences.
- Factors related to tablet manufacturing
- The amount and type of binder can affect the hardness, disintegration, and dissolution of tablets.
- The method of granulation, granule size, and size distribution can affect tablet dissolution.
- The concentration and type of disintegrants used, as well as the method of their addition, can affect disintegration and dissolution.
- Compression load can influence density, porosity, hardness, disintegration, and dissolution of tablets.
- Factors related to method of dissolution study
- Composition of the dissolution medium, pH, ionic strength, viscosity.
- Type of dissolution equipment.
- Temperature of the medium
- Volume of dissolution medium
- Intensity of agitation
- Sink or nonsink conditions (under a sink condition, the concentration of the drug should not exceed 10 – 15 % of its maximum solubility in the dissolution medium in use).
- Sensitivity of analytical method used to determine drug concentration in the release medium.
Non-Pharmacopoeial or Non-Official Tests
These are tests that are performed on tablets and which are not listed in official compendia and concern a variety of quality attributes that need to be evaluated, such as the porosity of tablets, hardness or crushing strength test, friability test, tensile strength determination, thickness test etc.
Some of these tests have no officially set limits for acceptance or rejection and thus may vary from manufacturer to manufacturer and from formulation to formulation.
Crushing strength and friability appeared in the 2001 Edition of British Pharmacopoeia (Appendix A324). There was however no definite set limits. The two tests are, therefore, here considered under non-pharmacopoeial tests.
Tablet Hardness or Crushing Strength Test
This measures the degree of force (in kilograms, pounds, or in arbitrary units) needed to fracture a tablet. Besides the concentration of binders used and the compression force, the hardness of a tablet depends on
- The characteristics of the granules to be compressed e.g., hardness and deformation under load.
- The type and concentration of lubricant used and
- The space between the upper and lower punches at the time of compression.
The crushing strength of tablets is usually checked using Monsanto or Stokes hardness tester, Strong-Cobb hardness tester and the Pfizer crushing strength tester. All are manually used. So, strain rate depends on the operator.

Hardness Tester
These equipment eliminate the operator variability encountered with manual hardness testers. Newer equipment with printers are also available.
A force of about 4 kg is considered the minimum requirement for a satisfactory tablet. Measurement is usually carried out using a minimum of ten tablets.
It has been found that a linear relationship exists between crushing strength and the logarithm of compressional force, except at high forces.
The strength of a cylindrical flat-faced tablet can be expressed as a tensile strength (Ts). This can be calculated as follows:
Where F is the force needed to fracture a cylindrical flat-faced tablet of thickness t along its diameter D.
Friability Test
This measures the resistance of tablets or granules to abrasion or fracture. The idea behind this test is to mimic the kind of forces, caused by phenomena such as collisions and sliding of tablets towards each other, which a tablet is subjected to during coating, packaging, handling, and shipping.
A minimum of 20 tablets are dedusted, weighed and subjected to a uniform tumbling motion for a specified time. They are then dedusted and reweighed.
The measure of abrasion/ friability loss is usually expressed as percentage loss in weight. It is calculated from the equation:
The test is rejected if any tablet caps, laminates or breaks up in course of the test. As a rule of thumb, a maximum weight loss of not more than 1% generally is considered acceptable for most pharmaceutical products. Values of up to 2% or above have been reported in direct compression formulations.
The friability of tablets may be influenced by moisture content. Chewable tablets show a high friability weight loss compared to conventional compressed tablets. A number of instruments are available for friability tests but the most popular and most reliable is the Roche Friabilator.

Dual drum friability tester
Tablet Thickness
Tablet thickness is determined by the diameter of the die, the amount of fill permitted to enter the die cavity, the compaction characteristics of the fill material, and the force or pressure applied during compression. To manufacture tablets of uniform thickness during and between batch productions for the same formulation, care must be exercised to employ the same factors of fill, die, and pressure.
The degree of pressure affects not only thickness but also hardness of the tablet; hardness is perhaps the more important criterion since it can affect disintegration and dissolution. Thus, for tablets of uniform thickness and hardness, it is doubly important to control pressure. Tablet thickness also becomes an important characteristic in packing operations and in counting of tablets using filling equipment which uses the uniform thickness of the tablets as a counting mechanism.
Tablet thickness is measured with a vernier caliper, thickness gauge or automated equipment (Automatic weight, hardness, thickness, and tablet diameter test instrument). The thickness of a tablet should be controlled within ±5% variation of a standard value depending on the size of the tablet.
Other non-pharmacopoeial tests include measurement of tablet diameter, porosity, liquid penetration, mechanical strength, and density.
Conclusion
Quality control of tablets involves various tests which require keen attention. To ensure that established product quality standards are met, these tests must be performed during production (in-process controls) and verified after the production of each batch.
References
- Allen L. V and Ansel H. C. (2014). Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott Williams and Wilkins.
- Dash, A. K., Singh, S. and Tolman, J. (2014). Pharmaceutics – Basic Principles and Application to Pharmacy Practice.USA: Academic Press.
- Felton, L.A. (2012). Remington Essentials of Pharmaceutics.UK: Pharmaceutical Press.
- Ghosh, T. K. and Jasti, B. R. (2005). Theory and Practice of Contemporary Pharmaceutics. USA: CRC Press LLC.
- Ofoefule, S. I. (2002). Textbook of Pharmaceutical Technology and Industrial Pharmacy.Nigeria: Samakin (Nig) Enterprise.
- Shayne, C. G. (2008). Pharmaceutical Manufacturing Handbook: Production and Processes.New Jersey: John Wiley & Sons, Inc.